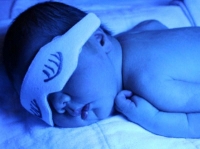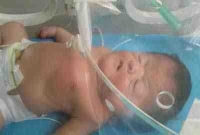
Caso Clínico: Se reporta un caso de recién nacido masculino, de 40 semanas más dos días de edad gestacional, con las características fenotípicas del Síndrome de Apert.
Discusión: Las características más relevantes del Síndrome de Apert radican en la fusión de algunos huesos del cráneo, manos y pies, dándole al paciente un aspecto característico. Es causado por mutaciones en el gen del receptor del factor de crecimiento fibroblástico tipo 2, localizado en el brazo largo del cromosoma 10. Su tratamiento es sobre todo quirúrgico-paliativo.
INTRODUCCIÓN
La craneosinostosis es el cierre prematuro de una o más suturas craneales, lo que ocasiona una alteración de la forma y configuración del cráneo. Puede ser un defecto aislado o un componente de uno de los numerosos síndromes craneosinostósicos que existen, entre los que se encuentran la Acrocefalosindactilia Tipo I o Síndrome Apert. (1)
El Síndrome de Apert, también conocido como acrocefalosindactilia tipo I, es un desorden congénito caracterizado por craneosinostosis coronal, sindactilia simétrica en las cuatro extremidades y malformaciones craneofaciales. El patrón de herencia es autosómica dominante y se considera que la mayoría de los casos de esta enfermedad surgen de forma esporádica. (2)
En cuanto a la frecuencia en la aparición del Síndrome de Apert es de aproximadamente 1 caso de cada 160.000 nacidos, con una distribución equitativa entre mujeres y hombre. Los asiáticos tienen el predominio más alto y los hispanos el más bajo. Estudios señalan que la aparición de esta patología es más frecuente entre los hijos de pareja de edad avanzada. (3)
Se ha demostrado el efecto de la edad paterna avanzada en el desarrollo de la enfermedad. (1)
Este trabajo se realiza para describir un caso clínico en el cual un recién nacido presenta la expresión fenotípica del Síndrome de Apert.
REPORTE DE UN CASO
Recién nacido de sexo masculino. Hijo de madre de 24 años de edad y padre de 35 años de edad. Producto de III Gesta I vivo I aborto, embarazo controlado (3 consultas públicas y 8 privadas) que no sugerían ningún tipo de malformación congénita, obtenido por parto eutócico simple a las 40 semanas más 2 días por FUR, líquido claro con grumos. Madre refiere leucorrea e infección del tracto urinario durante el II y III trimestre de embarazo. Al nacimiento se encuentra un peso de 3,800 gramos, talla de 52cm, circunferencia cefálica de 35,5cm y APGAR 7/8 puntos.
Al examen físico se evidencia craneosinostosis, exoftalmos leve, hipoplasia del tercio medio de la cara, pabellones auriculares de implantación baja, paladar hendido, cuello corto, sindáctila simétrica que compromete todos los dedos, espina bífida oculta, como se muestra en las figuras 1, 2 y 3. Ver Figura 1, 2 y 3.
En su evolución intrahospitalaria presentó Silverman de 8 puntos ameritando fluidoterapia, hidrocortisona y aminofilina. Falleciendo a las 24 horas de su nacimiento.
Paraclínicos: Cuenta Blanca: 14.600 Granulocitos: 83,1% Linfocitos: 10,4% Hemoglobina: 16,2gr/dl Plaquetas: 186.000 Glicemia: 141 VSG: 2 PCR: 6,0
DISCUSIÓN
Baumgartner hace las primeras menciones acerca de este defecto genético incluido dentro del amplio grupo de las anomalías craneofaciales en 1842, seguido por Wheaton en 1894; sin embargo, es Apert en 1906 quien lo describe y publica en detalle, razón por la cual a este síndrome se le conoce con su nombre. (4, 5)
La enfermedad se caracteriza por cierre precoz de las suturas craneales: sagital, coronal, escamosa y lambdoidea, con la aparición subsecuente de craneosinostosis; lo que conlleva a un crecimiento asimétrico de la cabeza. (6)
En la cara se observa: hipoplasia antero-posterior del tercio medio de la cara, hipoplasia del reborde orbitario dando la impresión de proptosis de los globos oculares, asimetría facial, puente nasal hundido, hipertelorismo, mandíbula prominente como efecto de la hipoplasia del tercio medio, fisuras palpebrales antimongoloides, pabellones auriculares grandes y por lo general de implantación baja. En cuanto a los maxilares se observa un maxilar superior con paladar ojival; pudiendo presentarse fisura palatina o úvula bífida, la mandíbula aparece en relación clase III con respecto al maxilar superior. (3)
Adicionalmente, se presenta la sindáctila de los dedos 2, 3 y 4, los cuales pueden aparecer soldados al pulgar y al quinto dedo y dar la apariencia de manopla o manos en mitón, defectos que pueden observarse en los pies y que reducen la flexibilidad de estos. La unión de los dedos puede estar confinada solamente a los tejidos blandos o involucrar también la parte ósea. Las extremidades superiores se encuentran acortadas con aplasia o anquilosis de algunas articulaciones, especialmente de hombros, codos y cadera. (3, 6)
La afectación de las manos clínicamente se clasifica según su severidad en tres tipos (7): el tipo 1 presenta sindactilia del 2º- 3º y 4º dedos con el 1º y 5º separados; en el tipo 2 la fusión afecta al 2º-3º-4º y 5º dedos con el 1º separado, dando a la palma de la mano una forma de cuchara; en el tipo 3 la sindactilia es completa, afectando a los 5 dedos. El tipo más frecuente de mano en el Síndrome de Apert es el tipo 1. La clasificación de la sindactilia de los pies es similar a los tipos observados en las manos, aunque, al contrario de lo que ocurre en las manos, las uñas de los pies están al menos parcialmente segmentadas y la fusión de las falanges distales que se observa en las manos no ocurre en los pies.
En la piel se presenta hiperhidrosis generalizada y acné vulgaris en cara, pecho, espalda y miembros superiores; también se observa un exceso de arrugas en la piel que cubre la frente la cual es debido a la hipoplasia ósea del hueso frontal. Existen algunas afecciones relacionadas con el Síndrome de Apert como lo son defectos cardiovasculares, atresia pulmonar, ducto arterial permanente, fistula traqueoesofágica, estenosis pilórica, riñones poliquísticos, infecciones otológicas y apnea del sueño. Se encuentra afectada la flexibilidad de la columna cervical, debido a la fusión presente en las vertebras cervicales C5- C6. (3)
Es necesario establecer diagnóstico diferencial del Síndrome de Apert con otros síndromes que presentan craneosinostosis (Tabla 1) (1).
Desde el punto de vista genético, es aceptado que el origen del Síndrome de Apert es una mutación puntual esporádica en la gran mayoría de los casos, que produce una ganancia en la función del receptor 2 del factor de crecimiento de los fibroblastos. Esta mutación se encuentra en el cromosoma l0q26 y consiste en la sustitución en dos codones adyacentes, en las posiciones 755TCG que codifica para serina y 758CCT que codifica para prolina en el FGFR2; de modo que se sustituye una C por una G y se expresa de la siguiente manera: TCG→TGG y CCT→CGT, codificando las siguientes proteínas: Ser252Trp y Pro253Arg. (8, 9)
En cuanto al diagnóstico prenatal del Síndrome de Apert, la ecografía tridimensional introducida en la década de 1980 suministra una ayuda para examinar el útero materno en ángulos diferentes, lo que permite conocer los defectos estructurales del cráneo, cara y extremidades del feto. En centros de alta tecnología de los países desarrollados se pueden observar, mediante un ultrasonido a partir de la semana 20 de gestación, defectos craneales. (10)
El tratamiento que se encuentra disponible es sobre todo quirúrgico-paliativo, se basa principalmente en separar las sinostosis presentes en el cráneo y las sindactilias en los miembros pélvicos y torácicos.
Existen líneas de investigación sobre opciones de tratamiento médico. Una de estas es la proteína Noggin; dicha proteína antagoniza a la proteína ósea morfogenética tipo 4 (BMP4 por sus siglas en inglés). La proteína Noggin previene el cierre de las suturas craneales y su regulación se encuentra a la baja en el Síndrome de Apert.
También se tiene la línea del calphostin C, que es un inhibidor de la proteincinasa C, elemento crucial en la señalización anómala en el Síndrome de Apert. El calphostin actúa mediante la modificación covalente del dominio regulatorio de la unión lipídica de la proteincinasa C, inhibiendo el cierre prematuro en las suturas craneales en modelos animales. (9)
Figura 2. Craneosinostosis y cuello corto en recién nacido con características fenotípicas del síndrome de Apert.

Síndrome de Apert. Craneosinostosis. Cuello corto
Fuente: propia del autor.
Figura 3. Sindactilia en pies del recién nacido con características fenotípicas del síndrome de Apert.

Síndrome de Apert. Sindactilia
Fuente: propia del autor.
Tabla 1: Diagnósticos diferenciales del Síndrome de Apert.
Síndrome: Apert
Gen y Herencia: FGFR2
Locus 10q25-q26
AD
Características craneofaciales: Turribraquicefalia, hipoplasia del tercio medio de moderada a severa.
Características extremidades: Sindactilia simétrica (manos y pies), acortamiento rizomélico, anquilosis de codos. No hay polidactilia.
Síndrome: Crouzon
Gen y Herencia: FGFR2 (diferentes partes excepto exón B).
Locus: 10q25-q26
AD
Características craneofaciales: Craneosinostosis coronal, exoftalmos, hipertelorismo, hipoplasia del tercio medio facial, nariz en “pico”, orejas de implantación baja.
Características extremidades: Ninguna alteración.
Síndrome: Carpenter
Gen y Herencia: RAB23
Locus: 6p12.1-q12
AD
Características craneofaciales: Craneosinostosis, atrofia óptica, fisuras palpebrales antimongoloides, orejas grandes y de implantación baja.
Características extremidades: Anomalías de los dedos de las manos y pies: braquidactilia, polidactilia, sindactilia, clinodactilia.
Síndrome: Saether- Chotzen
Gen y Herencia: TWIST1 (se han registrado algunos con mutación en FGFR2).
Locus: 7p
AD
Características craneofaciales: Braquicefalia, sinostosis coronal bilateral asimétrica, hipertelorismo, ptosis palpebral, menos frecuentemente hendidura palatina.
Características extremidades: Anomalías de los dedos de las manos y pies: braquidactilia y sindactilia parcial.
Síndrome: Pfeifer Tipo II
Gen y Herencia: FGFR1
Locus: 8p11.2-p11.1
FGFR2
Locus: 10q26
AD
Características craneofaciales: Craneosinostosis en cráneo en forma de trébol asimetría craneofacial, hipoplasia maxilar, hipertelorismo, ptosis palpebral, estrabismo, paladar ojival.
Características extremidades: Pulgares fusionados y anchos, braquidactilia, anquilosis de codos y rodillas, polidactilia.
REFERENCIAS
1. Vara, O., Milián, R., Piloña, S. y Rodríguez, J. Síndrome de Apert. Presentación de un caso neonatal. Rev Ciencias Médicas [revista en la internet]. 2006 nov [citado 2015 Feb 26]; 10(1): 51-60. Disponible en: http://scielo.sld.cu/scielo.php?pid=S1561-31942006000100006&script=sci_arttext
2. Ramírez, D., Saldarriaga, W., Pachajoa, H. y Isaza, C. Síndrome de Apert, una aproximación para un diagnóstico clínico. Reporte de caso. Salud Uninorte. [revista en la internet]. 2010 [citado 2015 Feb 26]; 26(1): 165-169. Disponible en: http://www.redalyc.org/articulo.oa?id=81715089016
3. Villarroel, A., Hochstatter, E. y Claustro, R. Síndrome de Apert (Acrocefalosindactilia). Gav Med Bol [revista en la internet]. 2007 [citado 2015 Feb 28]; 30(1): 58-62. Disponible en: http://www.scielo.org.bo/scielo.php?pid=S1012-29662007000100011&script=sci_arttext
4. Papp, E. Síndrome de Apert (Acrocefalosindactilia). Presentación de dos casos clínicos. Acta odontolog. Venez [revista en la internet]. 1999 Dic [citado 2015 Mar 01]; 37(3): 163-167. Disponible en: http://www.scielo.org.ve/scielo.php?pid=s0001-63651999000300030&script=sci_arttext
5. González, A., del Cerro, L., Álvarez, J., Warner, O., Santana, E. y Cardet, M. Síndrome de Apert. Reporte de un caso. Rev Cubana Genet Comunit [revista en la internet]. 2013 [citado 2015 Mar 02]; 7(2): 42-46. Disponible en: http://bvs.sld.cu/revistas/rcgc/v7n2/080213.pdf
6. Carro, E. y Fernández, L. Síndrome de Apert. Presentación de un caso. Rev Cubana Pediatr [revista en la internet]. 2005 Dic [citado 2015 Mar 03]; 77(3-4). Disponible en: http://scielo.sld.cu/scielo.php?pid=S0034-75312005000300009&script=sci_arttext
7. Carrera, A., Martínez, M., Marco, J., Paisán, L., Cárdenas, A., Nieto, C., et al. Síndrome de Apert: análisis clínico – epidemiológico de una serie consecutiva de casos en España. An Esp Pediatr [revista en la internet]. 1999 [citado 2015 Mar 04]; 51(6): 667-676. Disponible en: https://www.aeped.es/sites/default/files/anales/51-6-13.pdf
8. Atuasta, E., Balpardo, J., Echeverri, J., Estrada, C., López, D. y Roca, M. Síndrome de Apert esporádico. Reporte de un caso. Rev Mex Pediatr [revista en la internet]. 2009 [citado 2015 Mar 04]; 76(3): 121-123. Disponible en: http://www.medigraphic.com/pdfs/pediat/sp-2009/sp093d.pdf
9. Reséndiz, I. y Nava, E. Síndrome de Apert. Acta Med [revista en la internet]. 2013 [citado 2015 Mar 06]; 11(4): 173-179. Disponible en: http://www.medigraphic.com/pdfs/actmed/am-2013/am134b.pdf
10. Urdaneta, E., Vargas, Y., Urdaneta, A., Valero, J., Alviárez, L. y Contreras, P. Síndrome de Apert con agenesia renal, ¿una rara ocasión?. Rev Mex Pediatr [revista en la internet]. 2014 [citado 2015 Mar 07]; 81(1): 18-21. Disponible en: http://www.medigraphic.com/pdfs/pediat/sp-2014/sp141e.pdf